The thalassemia; are a set of hereditary hemolytic anemias that are caused by synthesis of either the alpha or beta chains that make up the hemoglobin, molecule. The reduction of a particular chain leads to the imbalance of globin chain synthesis and subsequently a distortion in the α:β ratio. As a result, unpaired globin chains produce insoluble tetramers that precipitate in the cell and cause damage to membranes.
The red cells are susceptible to premature red cell destruction by the reticuloendothelial system in the bone marrow, liver, and spleen.
In β thalassemia, there is an impairment of β-chain production that leads to an excess of alpha chains. These disorders are typically diagnosed several months after birth because the presence of β-chain is only important postnatally when it would normally replace the γ chain as the major non- α chain. There are multiple mutations that can lead to β-thalassemia. Almost any point mutation that causes a decrease in synthesis of mRNA or protein can cause this disease. Β-thalassemia is essentially an AR disorder seen more commonly among patients of Mediterranean descent, as well as Asians and Africans.
Sign and symptoms
Beta thalassemia minor – is clinically asymptomatic.
Beta thalassemia major – presents with symptoms of severe anemia. Patients are jaundiced, and leg ulcers and cholelithiasis occur. Splenomegaly is common, and the spleen may be several times its normal size.
Causes of poor growth in β thalassemia major
- Inadequate transfusion
- Hypogonadism
- Decreased growth hormone secretion/hypopituitarism
- Decreased growth hormone action due to receptor abnormalities
- Hypothyroidism, diabetes mellitus
- Adrenal hypo secretion
- Bone disorder
- Delayed puberty
- Trace element deficiency
- Intensive chelation
Diagnosis of beta thalassemia
Quantitative hemoglobin studies are used for routine clinical diagnosis. When a test shows elevated HbA₂, beta thalassemia is indicated. In β thalassemia major, HbF is usually increased, up to 90 percent.
In beta thalassemia major, X-ray findings are characteristic of chronic bone marrow hyperactivity. The skull and long bones are thinned. And the marrow space is widened. Tests show microcytic RBCs of varied size, with low hemoglobin. There are increased erythropoietin levels and, often, an overload of iron.
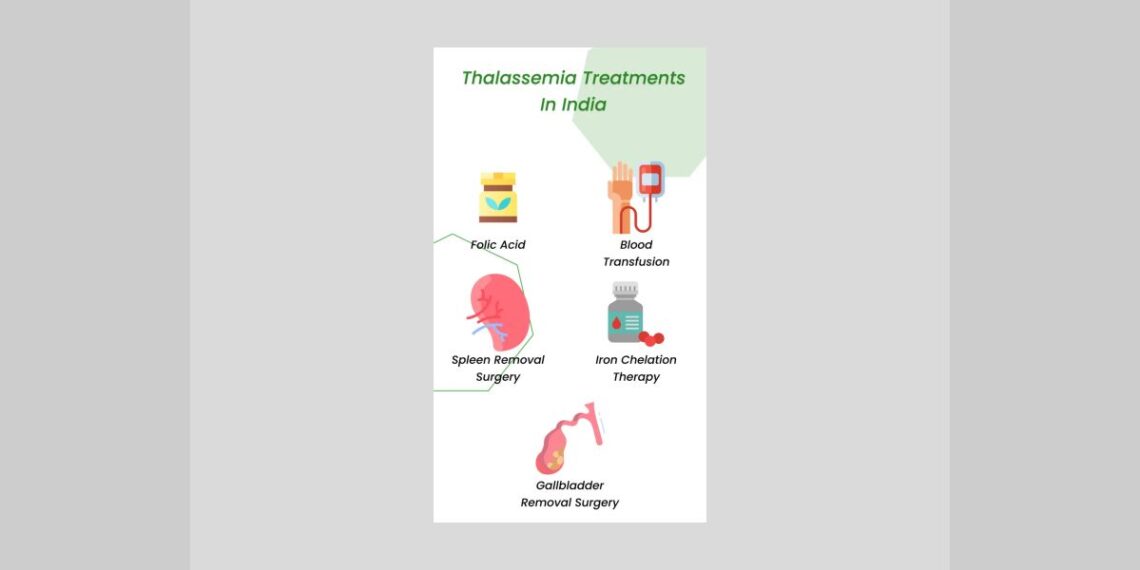
Homeopathic treatment for beta thalassemia symptoms
Homeopathy is one of the most popular holistic systems of medicine. The selection of remedy is based upon the theory of individualization and symptoms similarity by using holistic approach. This is the only way through which a state of complete health can be regained by removing all the sign and symptoms from which the patient is suffering. The aim of homeopathy is not only to treat beta thalassemia symptoms but to address its underlying cause and individual susceptibility. As far as therapeutic medication is concerned, several remedies are available to treat beta thalassemia symptoms that can be selected on the basis of cause, sensations and modalities of the complaints. For individualized remedy selection and treatment, the patient should consult a qualified homeopathic doctor in person.
Beta thalassemia may be the most best-known type of thalassemia and is also called Cooley’s anemia. It is caused by a change in the gene for the beta globin component of hemoglobin. Beta thalassemia causes variable anemia that can range from moderate to severe, depending in part on the exact genetic change underlying the disease. Beta thalassemia can be classified based on clinical symptoms. Beta thalassemia major usually causes severe anemia that can occur within months after birth. If left untreated, severe anemia can result in insufficient growth and development, as well as other common physical complications that can lead to a dramatically decreased life-expectancy. Fortunately, in developed countries beta thalassemia is usually identified by screening in the newborn period, before symptoms have developed. Children who are identified early can be started on ongoing blood transfusion therapy as needed. Although transfusion therapy prevents many of the complications of severe anemia, the body is unable to eliminate the excess iron contained in the transfused blood. Over time, the excess iron deposits in tissues and organs, resulting in damage and organ failure. Another medication must be administered to help the body eliminate the excess iron and prevent iron-over-load complications. Beta thalassemia intermedia describes the disease in individuals who have moderate anemia that only requires blood transfusions intermittently, if at all.