
Degree of Response to Homeopathic Potencies Correlates with Dipole Moment Size in Molecular Detectors: Implications for Understanding the Fundamental Nature of Serially Diluted and Succussed Solutions
DiagnOx Laboratory, Cherwell Innovation Centre, Upper Heyford, Oxon, United Kingdom
CC-BY-NC-ND 4.0 · Homeopathy 2018; 107(01): 019-031
DOI: 10.1055/s-0037-1617448
Original Research Article
The Faculty of Homeopathy
OPEN ACCESS
Abstract
Background The use of solvatochromic dyes to investigate homeopathic potencies holds out the promise of understanding the nature of serially succussed and diluted solutions at a fundamental physicochemical level. Recent studies have shown that a range of different dyes interact with potencies and, moreover, the nature of the interaction is beginning to allow certain specific characteristics of potencies to be delineated.
Aims and Methods The study reported in this article takes previous investigations further and aims to understand more about the nature of the interaction between potencies and solvatochromic dyes. To this end, the UV-visible spectra of a wide range of potential detectors of potencies have been examined using methodologies previously described.
Results Results presented demonstrate that solvatochromic dyes are a sub-group of a larger class of compounds capable of demonstrating interactions with potencies. In particular, amino acids containing an aromatic bridge also show marked optical changes in the presence of potencies. Several specific features of molecular detectors can now be shown to be necessary for significant interactions with homeopathic potencies. These include systems with a large dipole moment, electron delocalisation, polarizability and molecular rigidity.
Conclusions Analysis of the optical changes occurring on interaction with potencies suggests that in all cases potencies increase the polarity of molecular detectors to a degree that correlates with the size of the compound’s permanent or ground dipole moment. These results can be explained by inferring that potencies themselves have polarity. Possible candidates for the identity of potencies, based on these and previously reported results, are discussed.
Keywords
solvatochromic dyes – aromatic bridged amino acids – molecular detectors – dipole moments – homeopathic potencies
Introduction
Previous studies have demonstrated that solvatochromic dyes show significant changes in their UV-visible (US-vis) spectra under a range of conditions in the presence of homeopathic potencies.[1] [2] The approach taken in the current article has been to extend those investigations to include compounds that are not strictly solvatochromic but embody some crucial components of solvatochromic compounds in an attempt to further understand the molecular features necessary for dye–potency interaction. The methodology employed has been one in which a substantial number of aromatic compounds have been screened, the only requirements of the compounds tested being water solubility and an electron delocalised bridge between two polar groups. Surprisingly, it has been found that π-conjugated zwitterions respond to serially-diluted and succussed solutions. This discovery has revealed the existence of a large class of molecular detectors, which are in some ways superior to solvatochromic dyes for investigating potencies. For instance, π-conjugated zwitterions exhibit responses to potencies which, in one case, is the largest so far seen, and together with their relatively easy availability and potential for endless variations of structure, means specific aspects of the potency–dye interaction can be teased apart in some detail. As a consequence, several structural features necessary in order for compounds to be molecular detectors can now be delineated, and in turn several inferences can be made about the fundamental physicochemical nature of potencies.
Materials and Methods
Experimental Protocol
Experimental protocol is essentially as described previously.[1] [2] However, some minor improvements have been made and these are shown in [Fig. 1]. On obtaining potency and control solutions in 90% ethanol from the pharmacy, a 100-fold dilution was performed into reverse osmosis water (ROW) using standard amber moulded glass bottles from the same manufacturing batch. Exact material compatibility in terms of any leachates was established by inductively coupled plasma optical emission spectrometry (ICP-OES) at this stage. A further 100-fold dilution of each solution was then made into high density polyethylene (HDPE) bottles. These bottles were then stored separated by a minimum of at least 1.5 m.
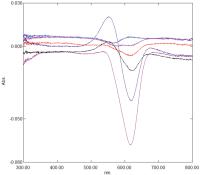
Fig. 1 Experimental protocol employed in this study (see text and Materials and Methods for details). HDPE, high density polyethylene.
Assays involved taking 50 μL of each solution and adding these aliquots to 2.95 mL of pre-prepared dye solution. Both control–dye and potency–dye solutions were then placed in black film canisters as described previously.[2] Difference spectra were run at intervals up to a maximum of 20 days. A total dilution of (100 × 100 × 60), or 600,000-fold, therefore occurs between solutions obtained from the pharmacy and solutions used for assays. Furthermore, as control and potency solutions approached depletion, ROW was added to both HDPE containers to replenish stocks. Over the course of the current studies both solutions have been replenished several times, resulting in a further c.100-fold dilution of the potency solution without any diminution of its effectiveness.
Assays have also been performed in which samples from the preceding amber molded glass bottles have been used, and no difference has been observed from results obtained from taking samples from the following HDPE bottles ([Fig. 1]). This indicates that leachates from amber molded glass bottles have no effect on results. The protocol outlined in [Fig. 1] has, therefore, been used as a precaution rather than as a necessity.
Unless otherwise stated Glycerol 50M has been used throughout this study. This has allowed comparisons to be made between all dyes, both in this study and in previous studies. At infrequent intervals, different potencies of Glycerol and potencies of other homeopathic medicines have been tested on the molecular detectors described in this article to ensure the methodology is not somehow specific to Glycerol 50M.
Reagents
6-amino-2-naphthoic acid (ANA), 4-aminobenzoic acid (ABA), 4′-amino-[1,1′-biphenyl]-4-carboxylic acid (ABPA), methylene violet (Bernthsen) (MV), coumarin 343 (C343), β-cyclodextrin (β-CD), cucurbit[7]uril (CB7), citric acid/sodium citrate, sodium dihydrogen phosphate/disodium hydrogen phosphate, boric acid/sodium borate and sodium N-cyclohexyl-3-aminopropanesulfonate (CAPS) were obtained from Sigma Aldrich UK and were of the highest purity available.
5, 6-diamino-naphthalene-1, 3-disulfonic acid (DANDSA) was obtained from Molekula, UK.
4-pyridinium phenolate (4PP) was synthesised and provided by WuXi App Tec (Hong Kong) Ltd. Structure and purity were confirmed by NMR.
The provenances of Brooker’s merocyanine (BM), bis-dimethylaminofuchsone (BDF), phenol blue (PB), and 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate (ET33) are as stated previously.[1] [2]
ROW was used throughout this study and had a resistivity of 15MΩcm (checked daily).
Disposable high purity UV-transparent cuvettes (Brand GmbH) with stoppers were used throughout and are described previously.[1] Disposable four-sided optically transparent fluorescence cuvettes (Brand GmbH) made of the same material as the UV cuvettes, with stoppers, were used to record fluorescence spectra.
Solution Storage
As in previous studies [1] [2] dye solutions were made and stored in HDPE bottles and allowed to equilibrate overnight before use. All dye solutions were stored in the dark as a precaution against light-induced degradation. This was deemed unnecessary but continued as a practice to ensure compatibility with previous studies.
Dyes were made up in buffers at concentrations sufficient to give an absorbance of between 0.5 and 1.5. Buffer solutions in which dye was dissolved were at a concentration of 20 mM throughout.
Homeopathic Potencies and Control Solutions
Glycerol 50M along with other potencies of glycerol were obtained from Helios Homeopathy Ltd, Tunbridge Wells, UK. All the results presented in this study were performed with Glycerol 50M.
Thirty microlitres of potency (in 90% ethanol) was diluted into 3.0 mL of ROW in an amber moulded glass bottle provided by the Homeopathic Supply Company Ltd, Bodham, UK. This ‘diluted’ aqueous potency was then further ‘diluted’ by transferring 30 μL into 3 mL of ROW in a 5 mL HDPE bottle. This final ‘HDPE’ potency solution was then used in assays ([Fig. 1]).
Control solutions were either un-medicated and un-succussed 90% ethanol obtained from Helios Homeopathy Ltd and diluted 100-fold as above into amber moulded glass bottles from the same batch as that used for potency dilutions, or control solutions consisted simply of ROW added to amber molded glass bottles from the same batch. As with potency solutions, a further 100-fold ‘dilution’ was performed into ROW in a 5 mL HDPE bottle, and this solution was used in assays ([Fig. 1]).
Final control and potency solutions in HDPE bottles were stored separated by a minimum of at least 1.5 m at room temperature in black plastic film canisters (Geo-Versand, GmbH, Germany).
Leachates from both potency and control bottles prior to dilution in HDPE bottles were analyzed by ICP-OES (Oxford-Analytical Ltd, Bicester, UK) and found to be at the same (<3μM) level for all elements tested (Ca, Mg, Si, K, Na, B, Fe). Dilution into ROW in HDPE bottles would then be expected to dilute those leachates to a < 0.03 μM level. A further 60-fold dilution occurs on addition of potency or controls to assay solutions.
Instrumentation
UV-vis spectra were recorded on a Shimadzu UV-2600 double-beam spectrophotometer.
Fluorescence spectra were recorded on a Shimadzu RF-6000 spectrofluorophotometer. Buffers were prepared using a Hanna pH210 microprocessor pH meter.
Experimental Procedures
Difference spectra were performed as follows. A total of 2.95 mL of buffered dye solution were pipetted into each of two Brand UV cuvettes with stoppers and the spectrophotometer set to zero across the wavelength range used for scanning (typically c.300–800 nm for solvatochromic dyes and 220/230–600 nm for aromatic bridged amino acids). Fifty microlitres of control solution was then added to the reference cuvette and 50 μL of potency solution added to the sample cuvette ([Fig. 1]). Cuvettes were inverted three times to mix and then scanned (t = 0). After the initial scan, both cuvettes were placed in separate black plastic film canisters (Geo-Versand, GmbH) to exclude all light and kept under these conditions between any subsequent scans. Scans were normally performed at t = 0 minutes, 10 minutes, 40 minutes, 100 minutes, c.200 minutes, c.6 hours, and c.12 hours after mixing. Subsequent scans were performed at intervals of days after mixing up to a maximum of 20 days.
Normal (non-difference) scans of dye solutions with potency or control solutions added were against ROW, which had been zeroed beforehand.
All assays were performed in 20 mM buffered solutions. Buffers used were citrate (pH 3–7), phosphate (pH 6–8), borate (pH 8–10), and CAPS (pH 10–11).
Fluorescence spectra were performed using Brand disposable four-sided optically transparent fluorescence cuvettes (Brand GmbH) made of the same material as Brand UV cuvettes. Separate fluorescence spectra were recorded of dye–control and dye–potency solutions transferred from assays in Brand UV cuvettes so spectra could be directly compared with UV-vis spectra at a set time.
Compounds Used in the Current Study
All of the compounds used in the current study are water soluble and this was one of the main criteria in selecting chromophoric reagents for their suitability. Structures of all compounds are given in [Fig. 2]. Six of the compounds— Methylene Violet (Bernthsen) (MV), Coumarin 343 (C343), Bis-dimethylaminofuchsone (BDF), 4-pyridinium phenolate (4PP), 2, 6-Dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate (ET33) and Brooker’s merocyanine (BM) are solvatochromic (see Appendix for definition); the first three being positively solvatochromic and the last three negatively solvatochromic. The other four compounds shown are essentially non-solvatochromic: that is their transition dipole moments are minimal.[3] They are members of a class of compounds comprising amino acids with an aromatic bridge. There exists very little in the literature on this type of compound. What is available pertains to 4-aminobenzoic acid.[4] [5] [6] Aromatic bridged amino acids consist of amino and carboxylic or sulfonic acid groups attached at opposite ends of a delocalised core of electrons. As with solvatochromic dyes, an electron or electron density is free to move between the two ends of the molecule under the influence of an appropriate stimulus. Unlike solvatochromic dyes, however, solvent polarity has little effect on the relative stability of the compounds’ ground and exited electronic states, and light does not cause a spatial movement of electrons. In principle, proton transfer can also occur with certain aromatic bridged amino acids, and this feature along with the other properties of this class of compound will be discussed in relation to the results obtained with potencies.
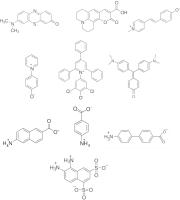
Fig. 2 Structures of molecular detectors used in this study. From top left: Methylene violet (Bernthsen) (MV), coumarin 343 (C343), Brooker’s merocyanine (BM), 4-pyridinium phenolate (4PP), 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate (ET33), Bis-dimethylaminofuchsone (BDF), 6-amino-2-naphthoic acid (ANA), 4-aminobenzoic acid (ABA), 4′-amino-[1,1′-biphenyl]-4-carboxylic acid (ABPA), 5, 6-diaminonaphthalene-1,3-disulfonic acid (DANDSA).
The amino acids with an aromatic bridge used in this study include 6-amino-2-naphthoic acid (ANA), 4-aminobenzoic acid (ABA), 5,6-diamino-naphthalene-1,3-disulfonic acid (DANDSA) and 4-amino-[1,1′-biphenyl]-4′-carboxylic acid (ABPA). While all four compounds respond to potencies with significant changes in their UV-vis spectra, results with DANDSA are of particular interest, as they provide the largest and most unusual spectral changes so far seen with any compounds, and provide insights into the potency–dye interaction which complement and add to those seen with the six solvatochromic dyes MV, C343, BM, 4PP, ET33 and BDF.
Results
N ≥ 5 for all spectra discussed below. Where spectra are shown, error bars have been omitted for clarity. [Table 1] provides pKa and dipole moment data for all the dyes tested along with their degree of response to Glycerol 50M and summarises the more detailed information given below.
Abbreviations: ABA, 4-aminobenzoic acid; ABPA, 4′-amino-[1,1′-biphenyl]-4-carboxylic acid; ANA, 6-amino-2-naphthoic acid; BDF, bis-dimethylaminofuchsone; BM, Brooker’s merocyanine; C343, coumarin C343; ET33, 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate; DANDSA, 5, 6-diamino-naphthalene-1,3-disulfonic acid; MV, methylene violet (Bernthsen); 4PP, 4-pyridinium phenolate; PB, phenol blue.
a pKa values determined spectroscopically (this study).
b Dipole moment values estimated according to established principles.[16]
c Percentage change in dye spectra under the influence of potency is a combination of steps one, two and three (see text for explanation).
Assays at pH Values ≈ dye pKas
Methylene Violet (Bernthsen)
[Fig. 3] shows a typical series of difference spectra of MV ± potency at intervals up to 11 days (50 μM dye, 20 mM citrate buffer pH 4.0). What is striking is the size of the spectral changes, constituting c. 7% of the total absorbance of the dye (OD = 1.0 at 614 nm). As with previous studies[1] [2] the difference spectrum is slow to appear but is then relatively stable over long time periods. The decrease at 614 nm is consistent with potency promoted protonation and loss of monomer. MV is conformationally inflexible ([Fig. 2]) and this may be a factor along with its high dipole moment (≥18D[7]) in conferring a high degree of response to potencies. Fluorescence studies have confirmed that potency promotes MV aggregation (fluorescence spectra show a decrease in fluorescence intensity in the presence of potency).[7] [8]

Fig. 3 Difference spectrum of 50 μM MV in 20 mM citrate buffer pH 4.0 with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 0, t = 10 minutes, t = 100 minutes, t = 260 minutes, t = 4 days and t = 11 days after mixing (see text for details). MV, methylene violet (Bernthsen).
Coumarin 343
Coumarin 343 is a structural analogue of MV and shows spectral differences very similar to the latter dye but with a decrease at 427 nm ([Fig. 4]). The difference spectra shown are of 50 μM dye in 20 mM phosphate buffer pH 7.0 ± potency. The changes observed are 4 to 5% of the total absorbance of the dye (1.6 at 427 nm). The inclusion of C343 in this study is important because of its smaller dipole moment relative to that of MV, although like MV, it is structurally inflexible. Difference spectra again are slow to develop and fluorescence studies show that potency promotes C343 aggregation.
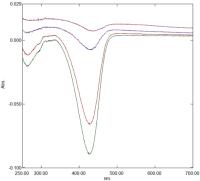
Fig. 4 Difference spectrum of 50 μM C343 in 20 mM phosphate buffer pH 7.0 with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 17 minutes, t = 150 minutes, t = 12 hours and t = 7 days after mixing (see text for details). C343, coumarin C343.
Brooker’s Merocyanine
[Fig. 5] shows difference spectra of 90 μM BM in 20 mM borate buffer pH 8.5 ± potency with 10 mM β-CD[9] present. Again large, slowly appearing, spectral changes are seen under the influence of potency, constituting finally some 8% of the total absorbance of BM at this pH (OD = 1.4 at 465 nm). Importantly, in the absence of β-CD, the spectral changes are considerably reduced and very variable. β-CD is a molecular encapsulator which has a two-fold effect. First, it prevents any aggregation of BM and second it rigidifies the dye molecule. Its Kassoc for BM is c.2 mM,[10] so BM is likely to be fully encapsulated at the concentration of β-CD used in the assays. The fact that difference spectra are so much larger and more stable with encapsulation suggests rigidification of BM improves its response to potency. BM is known to isomerise easily[11] and is, therefore, an intrinsically mobile molecule, which may preclude easy interaction with potency. The spectral changes seen in [Fig. 5] are consistent with potency-induced protonation of BM.

Fig. 5 Difference spectrum of 90 μM BM in 20 mM borate buffer pH 8.5 containing 10 mM β- cyclodextrin with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 0, t = 100 minutes, t = 200 minutes, t = 385 minutes, t = 12 hours and t = 14 days after mixing (see text for details). BM, Brooker’s merocyanine.
4-Pyridinium Phenolate
This negatively solvatochromic dye is the simplest example of the pyridinium phenolates, of which ET30 and ET33 have already been examined.[1] [Fig. 6] shows a series of difference spectra of 100 μM 4PP at pH 8.5 in 20 mM borate buffer ± potency over time. Changes are again slow to appear and constitute c.3% of the total absorbance of the dye (OD = 0.5 at 375 nm) at their maximum. As with MV and BM, the spectral changes seen are consistent with potency-induced protonation.[1] [12]
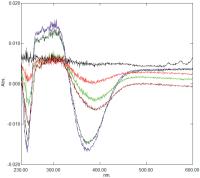
Fig. 6 Difference spectrum of 100 μM 4PP in 20 mM borate buffer pH 8.5 with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 0, t = 10 minutes, t = 40 minutes, t = 100 minutes, t = 12 hours and t = 2 days after mixing (see text for details). 4PP, 4-pyridinium phenolate.
6-Amino-2-Naphthoic Acid
Like DANDSA below, 6-Amino-2-Naphthoic Acid (ANA) is a π-conjugated amino acid and not solvatochromic. Nevertheless [Fig. 7] shows a series of spectra of 200 μM ANA in 20 mM citrate buffer pH 3.5 ± potency. The decreases at c.315 nm and 250 nm are consistent with slow protonation induced by potency. Spectral changes constitute c.3 to 4% of the total absorbance of ANA (OD = 0.8 at 315 nm).
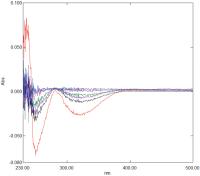
Fig. 7 Difference spectrum of 200 μM ANA in 20 mM citrate buffer pH 3.5 with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 0, t = 10minutes, t = 40 minutes, t = 100 minutes, t = 200 minutes and t = 4 days after mixing (see text for details). ANA, 6-amino-2-naphthoic acid.
5,6-Diamino-Naphthalene-1,3-Disulfonic Acid
[Fig. 8] shows a series of spectra obtained of 100 μM DANDSA in 20 mM citrate buffer pH 4.0 ± potency. Initial spectra reveal decreases at 405 and 268 nm, with increases at 343 nm and 250 nm. These changes are consistent with potency-induced protonation. Over longer time periods, new difference peaks appear at 415, 393, and 298/305 nm, all associated with dye aggregation. This conclusion is confirmed by fluorescence spectroscopy where fluorescence intensity decreases in the presence of potency. The total changes in absorbance of DANDSA in the presence of potency amount to 8 to 10% of overall absorbance (OD = 0.2 at 415 nm), meaning this compound is the most sensitive reporter so far discovered.
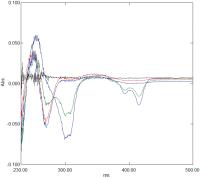
Fig. 8 Difference spectrum of 70 μM DANDSA in 20 mM citrate buffer pH 4.0 with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 0, t = 100 minutes, t = 220 minutes, t = 7 days and t = 18 days after mixing (see text for details). DANDSA, 5, 6-diamino-naphthalene-1,3-disulfonic acid.
DANDSA demonstrates the first clear evidence that potency is initially acting to change the pKa value of a molecular reporter, which is then followed by changes in aggregation levels, rather than by acting directly on aggregation levels. These results are discussed below in relation to a proposed common mechanism of action of potencies on all molecular reporters so far examined.
4-Aminobenzoic Acid
4-Aminobenzoic Acid (ABA) is the smallest and simplest molecule examined for the effects of potency. Surprisingly perhaps, despite its size, it also demonstrates changes in its UV spectrum in the presence of potency with a decrease in absorbance at c. 292 nm in 20 mM citrate buffer pH 3.5. This change constitutes c.1% of the total absorbance of ABA and is consistent with potency-induced protonation. It is significant that ANA, a molecule that differs from ABA only in the length of its aromatic bridge ([Fig. 2]), and consequently its dipole moment, should respond more strongly than ABA. The importance of this structural difference between ANA and ABA in relation to their ability to respond to potencies is discussed below.
4′-Amino-[1, 1′-Biphenyl]-4-Carboxylic Acid
4′-Amino-[1, 1′-Biphenyl]-4-Carboxylic Acid (ABPA) is a structural analogue of ANA in which the naphthalene ring is replaced by a biphenyl electron delocalised bridge. Difference spectra, ± potency in 20 mM citrate buffer pH 4.2 show a decrease at c.305 nm and an increase at c.270 nm, consistent with potency-induced protonation. Overall absorbance changes constitute c.2% of total absorbance. This lower number compared with that found for ANA may reflect the conformational mobility of ABPA compared with ANA, an issue already mentioned in relation to BM, and discussed in more detail below.
The above results from eight different molecular reporters demonstrate that potencies interact with a range of structural forms to produce significant spectral changes. Several compounds, and particularly DANDSA, have indicated that there are, however, at least two steps involved in the production of these spectral changes. The first involves potency-induced protonation. As solutions are buffered and it is known that ordinary pH indicators show no response to potencies,[2] together with the slow appearance of spectral changes over hours, this suggests some kind of electron density shift occurs across the molecules resulting in altered pKa values.[13] [14] This conclusion has already been deduced from results obtained with BDF in a previous study.[2] The current study has provided further evidence that this indeed may be the case. If potencies are producing a pKa change in molecular detectors, then a preceding step involving some kind of electron density movement across the molecules may well be the primary form of the interaction between potencies and molecular detectors.
This possibility can be tested in the following way. If assays are performed at pH values well away from the pKa value of compounds, then protonation/deprotonation is not possible, and the putative step two is silenced. If molecular encapsulators such as β-CD[9] or cucurbiturils[15] are added to solutions to prevent any aggregation of compounds, then step three is also silenced. Any spectral changes in the presence of potencies are then likely to be attributable to an earlier, possibly primary, step.
The following results pertain to assays performed at pH values >> pKa values and in the presence of molecular encapsulators with dyes MV, BDF, BM, 4PP and ET33. It should be noted here that only solvatochromic dyes are capable of showing sufficient spectral changes due to spatial electron movement and so amino acids with an aromatic bridge cannot be tested for this step directly.
Assays at pH Values >> Dye pKas
Positively Solvatochromic Dyes MV and BDF
[Fig. 9] shows difference spectra (right) obtained with 50 μM MV in 20 mM borate buffer pH 9.0/10 mM β-CD ± potency at t = 100 and 220 minutes. A peak at 629 nm is evident. The λmax of a control solution of MV in the same β-CD/borate buffer is at 617 nm (left) and this is attributable to monomer, with a shoulder at c.574 nm to dimer. The new peak at 629 nm in the presence of potency can, therefore, be confidently assigned to a form of monomer. Positively solvatochromic dyes display bathochromic shifts in their spectra with increasing stabilisation of the excited (more charged) state ([Fig. 10]). It seems reasonable to conclude, therefore, that potency is stabilising something similar to the excited state of MV in which the opposite ends of the molecule are becoming more formally charged.

Fig. 9 Difference spectra of MV in 20 mM borate buffer pH 9.0 containing 10 mM β-cyclodextrin with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette. Spectra correspond to t = 100 minutes and t = 220 minutes after mixing. Maxima are at 629 nm (right-hand curves). Control solution of MV in the same β-cyclodextrin buffer (left-hand curve). Maximum is at 617 nm. See text for details. Spectra are not to scale. MV, methylene violet (Bernthsen). Fig. 10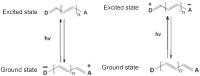 Potencies are postulated to interact with and stabilise the ground (more polar) state of negatively solvatochromic dyes (left) and the excited (more polar) state of positively solvatochromic dyes (right).
Potencies are postulated to interact with and stabilise the ground (more polar) state of negatively solvatochromic dyes (left) and the excited (more polar) state of positively solvatochromic dyes (right).
A comparable result to that with MV is seen with BDF in 20mM borate buffer pH 9.0/10 mM β-CD ± potency. In this case, a new peak appears at 583 nm compared with the λmax of a control solution of BDF in the same buffer which is at 567 nm. Again, potency seems to be stabilising a more polar form of BDF. This can only occur if an electron density movement has occurred toward the carbonyl moiety of BDF, as previously suggested may be happening.[2]
Negatively Solvatochromic Dyes BM, 4PP and ET33
[Fig. 11] shows a difference spectrum of 50 μM ET33 in 20 mM borate buffer pH 8.5/10 mM β-CD ± potency. While the differences are small, they nevertheless show a hypsochromic shift in the presence of potency with a decrease at 470 nm and an increase at 373 nm. The λmax of a control solution of ET33 in the same buffer is at 407 nm. In contrast to results seen with the positively solvatochromic dyes BDF and MV, potency is inducing a hypsochromic shift in the spectrum of ET33. Negatively solvatochromic dyes display hypsochromic shifts in their spectra with increasing stabilisation of their ground (more charged) state ([Fig. 10]). It seems, therefore, that in the presence of potency the ground state of ET33, which is already charged, is having its polarity increased even further.
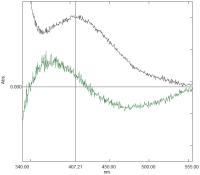
Fig. 11 Difference spectrum of ET33 in 20 mM borate buffer pH 8.5 containing 10 mM β- cyclodextrin with control added to the reference cuvette and Glycerol 50 M added to the sample cuvette showing a decrease at 470 nm and an increase at 373 nm (bottom curve). Spectrum corresponds to t = 210 minutes after mixing (see text for details). Control spectrum of ET33 in the same buffer containing 10 mM β-cyclodextrin shows an absorbance maximum at c.407 nm (top curve). Spectra are not to scale. ET33, 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate.
Similar results have been obtained with 4PP and BM. [Table 2] shows a summary of the results obtained with all five dyes. It would appear from these results that potency is preferentially interacting with, and intensifying, the charged forms of both positively and negatively solvatochromic dyes.
| Table 2
The effect of potency on dye spectra in the presence of β-cyclodextrin and at pH values >> the pKa of dyes. Left-hand column gives dye maxima in control solutions of buffer/β-cyclodextrin; the right-hand column shows the effect of potency |
||
| Dye | β-CD[c]/control | β-CD[c]/potency |
| BDF[a]
pH 9.0 |
λmax 567 nm | New peak at c.583 nm |
| MV[a]
pH 9.0 |
λmax 617 nm | New peak at c.629 nm |
| BM[b]
pH 11.0 |
λmax 456.5 nm | Decrease at c.495 nm
Increase at c.390 nm |
| 4PP[b]
pH 11.0 |
λmax 367 nm | Decrease at c.400 nm
Increase at c. 330 nm |
| ET33[b]
pH 8.5 |
λmax 406.5 nm | Decrease at c.470 nm
Increase at c.373 nm |
Abbreviations: β-CD, β-cyclodextrin; BDF, bis-dimethylaminofuchsone; BM, Brooker’s merocyanine; ET33, 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate; MV, methylene violet (Bernthsen); 4PP, 4-pyridinium phenolate.
a Positively solvatochromic dyes.
b Negatively solvatochromic dyes.
c β-cyclodextrin concentration 10 mM/buffer concentration 20 mM (pH 8.5 and 9.0 borate; pH 11.0 CAPS).
Discussion
The current study has considerably extended the range of compounds that respond to homeopathic potencies. Solvatochromic dyes now seem to be a sub-group of a larger class of compounds known as π-conjugated dipoles demonstrating interactions with serially diluted and succussed solutions. These include amino acids with an aromatic bridge (π-conjugated zwitterions). The presence of a large dipole moment, electron delocalisation, polarizability (the ability for electron density to shift across the molecule under an appropriate stimulus) and molecular rigidity seem to be general requirements in compounds for significant interactions with potencies to take place. Two particular compounds of this wider class which are readily available and provide significant spectroscopic responses to potencies are ANA and DANDSA, the latter demonstrating changes in its spectra of 8 to 10% over time.
[Fig. 12] shows a plot of percentage change in dye spectra versus dye dipole moment. Some uncertainty exists over the ground or permanent dipole moment size for several compounds used in this study as their values are not available in the literature, but reasonable estimates can be made according to established principles.[16] Despite these minor uncertainties in dipole moment size, the general trend is clear. The larger the size of the dipole moment of a compound, the larger the response to potencies appears to be. PB, examined previously,[1] has the smallest ground dipole moment and produces the smallest response. Conversely MV, BM, and DANDSA have the largest dipole moments and produce the largest responses. In addition, comparing ABA with ANA, where the only difference is the distance between charged moieties, and hence dipole moment, a threefold increase in response is seen.

Fig. 12 Plot of percentage change in dye spectra versus dye ground or permanent dipole moment for solvatochromic compounds (upper plot) and non-solvatochromic compounds (lower plot) used in this study. 4ABA, 4-Aminobenzoic acid; ABPA, 4′-amino-[1,1′-biphenyl]-4-carboxylic acid; ANA, 6-amino-2-naphthoic acid; β-CD, β-cyclodextrin; BDF, bis-dimethylaminofuchsone; BM, Brooker’s merocyanine; C343, Coumarin 343; DANDSA, 5, 6-diamino-naphthalene-1,3-disulfonic acid; MV, methylene violet (Bernthsen); PB, phenol blue; 4PP, 4-pyridinium phenolate.
The slow appearance of spectra ([Figs. 3] [4] [5] [6] [7] [8]) and the correlation between detector polarity and degree of response suggests some kind of synergistic process or resonant interaction taking place between detector and potency in which the polarity of both are gradually strengthened. The success and magnitude of such an interaction may well depend upon several factors (see below).
Several other insights emerge from [Fig. 12]. It is clear that in contrast to the ground or permanent dipole moment, the transition dipole moment (the difference between ground and excited states) is not an indicator of response to potencies and there does not appear to be any correlation. For instance, amino acids with an aromatic bridge such as ANA and DANDSA have negligible transition dipole moments, yet demonstrate responses. Pyridinium phenolates all have very large transition dipole moments of c. 20–22D[3] [17] yet display modest spectral changes ([Table 1]).
Dipole moment size may not be the only determinant underlying response to potencies, however. MV is a conformationally rigid molecule compared with, for example, 4PP, which has a similar dipole moment, and yet there exists a significant difference in the magnitude of their responses to potency. Furthermore, BM is conformationally mobile and produces modest and very variable responses, and yet on encapsulation, which renders it more rigid, response increases considerably. These results may indicate that molecular rigidity is required for potencies to effectively engage with molecular detectors. Additional evidence for this proposition comes from results with C343 where response to potencies is greater than for 4PP and ET33 ([Table 1]).
Using a range of compounds, both solvatochromic and non-solvatochromic, has not only demonstrated that specific molecular features (large dipole moment, electron delocalisation, polarizability and molecular rigidity) appear to be important for interactions with potencies to take place, but it has also revealed that several steps are involved in the production of spectral changes indicative of these interactions.
Results pooled from all 10 compounds reported in this study indicate that there are three steps in the interaction between potencies and molecular detectors that produce the spectral changes seen. While not all steps are separately observable in all compounds so far tested, it seems likely that these steps are a common feature given the number of overlaps between results. [Table 3] summarises results for all compounds investigated and reported herein.
Abbreviations: ABA, 4-aminobenzoic acid; ABPA, 4′-amino-[1,1′-biphenyl]-4-carboxylic acid; ANA, 6-amino-2-naphthoic acid; β -CD, β -cyclodextrin; BDF, bis-dimethylaminofuchsone; BM, Brooker’s merocyanine; ET33, 2, 6-dichloro-4-(2, 4, 6-triphenyl-pyridinium-1-yl)-phenolate; DANDSA, 5, 6-diamino-naphthalene-1,3-disulfonic acid; MV, methylene violet (Bernthsen); 4PP, 4-pyridinium phenolate.
a Not investigated.
b BDF pKa1 represents BDF + H+↔ BDF-H+ and pKa2 BDF + OH–↔ BDF-OH–.
c Relative aggregation levels observable by resonant light scattering (this study).
d Relative aggregation levels observable by fluorescence spectroscopy (this study).
e Relative aggregation levels observable by UV-vis and fluorescence spectroscopy (this study, see text for more details).
Step one appears to involve a primary interaction between potency and molecular detector, resulting in an electron density shift. This step is observable with positively and negatively solvatochromic dyes. Both are made more polar—the former having their excited (charged) state stabilised, and the latter having their ground (charged) state stabilised.
Step two follows on from step one and is a consequence of it. Any electron density shift in a delocalised system will cause a change in one or more of any ionisable groups attached to that system.[14] For all compounds assayed, changes in pKa values are seen. For amino acids with an aromatic bridge, this is the first observable step as these compounds do not have the spectral characteristics of solvatochromic dyes and hence step one is silent. While not solvatochromic, aromatic bridged amino acids, which are zwitterionic,[13] produce significant interactions with potencies. These results taken together suggest that potencies preferentially interact with polar species, rendering them more polar. Indeed, the more polar the detector, the more effect potency seems to have ([Fig. 12]).
Step three results from step two. Any change in pKa values and hence degree of protonation will affect aggregation levels, as the forces driving aggregation include ionic as well as hydrophobic interactions and hydrogen bonding.[18] Step three is most clearly separately observable with DANDSA, but is also apparent with compounds such as BDF and MV.
A final observation may have some relevance to the discussion that follows. A previous study has found that sustained light exposure inhibits the BDF–potency interaction, and continuous irradiation results in no observable spectroscopic difference between dye–control and dye–potency solutions.[2] This observation has now been extended to include 4PP, ET33, BM, MV and ANA. In all cases, no effect of potency is seen with any of the above compounds on continuous irradiation (usually at the absorbance maximum of the dye). Short exposure to light (i.e., during assay in the spectrophotometer) has little effect, so continuous exposure is necessary to fully inhibit the dye–potency interaction. In addition, any loss of spectroscopic differences from medium term light exposure to dye–potency solutions can be reversed by placing solutions in the dark.
Consequently, to avoid complications created by light exposure, and as described in the Materials and Methods, all dye–potency and dye–control incubations are performed in the dark and assays performed at intervals, with incubations being returned to black film canisters in between assays.
Curiously, however, irradiation of potency solutions themselves appears to have no deleterious effect, at least over the short term. In addition, all aromatic bridged amino acid–potency solutions are scanned down to at least 230 nm, and often 220 nm, when assayed. This is well into the UV region of the electromagnetic spectrum, and yet no obvious loss of potency strength has been observed. It may be that potencies are far more robust than previously thought and that any UV-induced inactivation is slow and requires multiple exposures.
Why potency solutions themselves appear to be immune to light exposure and yet dye–potency interactions are sensitive remains unclear at this stage. It is likely, however, to be saying something fundamental about the physico-chemical nature of potencies.
Returning to dipole moment size and degree of response in relation to the putative step one above, it would seem that the larger the dipole moment of molecular detectors, the larger the response to potencies. This implies that potencies require polarity to interact, and on interaction increase the polarity of detectors proportionately. This in turn suggests potencies themselves must have polarity to result in this kind of interaction, as has been put forward previously.[2] Any proposal as to the possible physico-chemical nature of potencies must, therefore, include polarity in the proposal.
Several well-known facts about potencies are perhaps worth reconsidering at this point.
The first is that potency solutions are prepared and stored as water–alcohol mixtures, often with the proportion of alcohol present as high as 90%.
Second, homeopathic potencies are dispensed most usually in the form of tablets. Both of these facts argue against water as being the source of potencies, but allow for the possibility that water, alcohol, and lactose may be carriers.
Third, potencies can be administered by olfaction. Indeed, Hahnemann and many of his colleagues and followers used this method.[19]
Finally, both trituration and succussion have a feature in common, and that is friction. Both grinding and vigorous shaking are forms of friction, or the action of one surface against another.
Combining the above observations–namely, that potencies have polarity, potencies are created by friction, potencies may be administered by olfaction, and water, alcohol, or lactose are unlikely to be the source of potencies (but may be carriers)–it is difficult to avoid the possibility that potencies might be some form of non-thermal plasma,[20] however improbable that may seem. Non-thermal plasmas are produced by friction. Indeed, both triboplasma (plasma produced by grinding)[21] and cavitation plasma (through violent shaking of solutions and bubble implosion) are well-documented phenomena.[22] Plasmas are composed of free ions and as such become highly polarised under the influence of electrical and magnetic fields.[20] If such plasmas were stable, they could be administered by olfaction. That plasmas emit light may also be relevant to the observation that some curious relationship exists between potency action and the necessity to keep light excluded from dye–potency solutions.
The improbability of the suggestion that potencies may be non-thermal plasmas carried by polar vehicles, such as water, alcohol and lactose, lies however in the transitory nature of, and the high levels of energy required to sustain plasmas for any length of time.[20] Nevertheless, there have been several proposals over the years, including recently,[23] that potencies and plasmas share much in common and that succussion does, momentarily, produce plasma. Clearly more would need to be done to strengthen or discount the possibility that potencies may be some form of non-thermal plasma.
Conclusions
The present study has demonstrated that a wide range of compounds under the general category of π-conjugated dipoles respond to homeopathic potencies. These include solvatochromic dyes as well as amino acids with an aromatic bridge (π-conjugated zwitterions), which carry formal charges at either end of their delocalised systems. This greatly extends the number of molecular detectors available and has provided valuable insights into the fundamental nature of potencies. Solvatochromic dyes now appear to be a sub-set of a much wider group of compounds sensitive to serially succussed and diluted solutions. The necessary requirements for sensitivity to potencies appear to be electron delocalised systems that have a large permanent or ground dipole moment, together with the ability of the system to be polarised, meaning their electron density is free to move spatially across the molecule under the influence of appropriate stimuli. The larger the permanent or ground dipole moment of such compounds, the more they are polarised in the presence of potencies. In addition, molecular rigidity appears to be an important structural component of molecular detectors and improves responsiveness further.
Amino acids with an aromatic bridge demonstrate significant responses to potencies, and results with these compounds, particularly DANDSA, have confirmed and extended those reported previously with solvatochromic dyes. The large dipole moments and molecular rigidity of these π-conjugated zwitterions appear to be responsible for their responses to potencies.
Using a combination of extensive screening of potential molecular detectors of different structures, molecular encapsulation with β-CD and assaying at pH values both near pKas of compounds, as well as at pH values far removed from their pKa values, has revealed that the generation of difference spectra proceeds in three steps. The first step appears to involve the primary interaction of potency and molecular detector, producing a shift in electron density across the molecule. This step can be detected using solvatochromic dyes, which are encapsulated with β-CD and assayed ± potency at pH values well away from the pKa value of the dye. Positively solvatochromic dyes exhibit a bathochromic shift in their spectra, indicating stabilisation of the dyes’ excited (and more polar) state, while negatively solvatochromic dyes exhibit a hypsochromic shift in their spectra, indicating stabilisation of the dyes’ ground (and more polar) state.
The second step can be detected in all compounds assayed at pH values ≈ pKa values and is the result of the first step. Electron density shift in step one results in a change in pKa values and protonation levels, which produce spectroscopic changes characteristic of each compound.
The third step can be most clearly seen with the molecular detector DANDSA owing to the very different spectra of aggregated and disaggregated material, and where a change in protonation levels leads much more slowly to enhanced aggregation of the compound in the presence of potency. The third step can also be discerned with MV and BDF, using a combination of UV-vis and fluorescence spectroscopy ([Table 3]).
Finally, it has been proposed that the possibility that potencies are some form of non-thermal plasma should at least be entertained, despite the obvious objections that such a proposal raises. The apparent polarity of potencies, their generation through friction, their storage in ethanol/water mixtures and on lactose, together with the observation that they can have their clinical effect through olfaction, argues against any kind of standard pharmaceutical formulation and points more towards an electromagnetic identity for potencies. The observation that dye–potency interactions are inhibited by light only further emphasises this possibility.
Appendix
Non-thermal plasma: A partially ionised gas at room temperature in which electrons are free and not bound to any atom or molecule. Plasmas exhibit many interesting properties including sensitivity to, and the generation of, electromagnetic fields, collective behaviours, dissipative structures, coherence and self-organisation. Plasmas also have electron oscillation frequencies.
π-conjugated dipole: A molecule in which there is a higher electron density at one end than the other and where both ends are connected by an electron bridge of delocalised or free electrons. A π-conjugated zwitterion is where the electron density disparity between each end of the molecule is such as to have become formal positive and negative charges. Electron density is free to move along the length of π-conjugated dipoles under the influence of appropriate stimuli, such as electromagnetic fields.
Solvatochromism: The ability of a chromophoric compound to change colour with a change in solvent polarity. This is due to a difference in the dipole moment between the ground and excited states of the chromophore and involves a spatial movement of electron density along the length of the molecule under the action of an appropriate stimulus such as light. Solvatochromic compounds are π-conjugated dipoles and as such are also sensitive to the presence of electromagnetic fields.
Conflict of Interest
No source of funding had any influence on the design, analysis, interpretation or outcome of the research contained within this manuscript, nor on the writing of the manuscript.
Acknowledgments
Funding for this work is gratefully acknowledged from The Homeopathy Research Institute, UK; Standard Homeopathic Company/Hylands, USA and The Tanner Trust, UK.
- References
- 1 Cartwright SJ. Solvatochromic dyes detect the presence of homeopathic potencies. Homeopathy 2016; 105: 55-65
- 2 Cartwright SJ. Interaction of homeopathic potencies with the water soluble solvatochromic dye bis-dimethylaminofuchsone. Part 1: pH studies. Homeopathy 2017; 106: 37-46
- 3 Reichardt C, Welton T. Solvent effects on the absorption spectra of organic compounds. In Solvents and Solvent Effects in Organic Chemistry. 4th ed. Weinheim: Wiley-VCH; 2011: 365-367
- 4 Thayer MP, McGuire C, Stennett EM. , et al. pH dependent spectral properties of para-aminobenzoic acid and its derivatives. Spectrochim Acta A Mol Biomol Spectrosc 2011; 84: 227-232
- 5 Halpern A, Ramachadran BR. The photophysics of ρ-aminobenzoic acid. Photochemistry and Photobiology 1995; 62: 686-691
- 6 Gainer A, Stevens JS, Suljoti E. , et al. The structure of ρ-aminobenzoic acid in water: studies combining UV-vis, NEXAFS and RIXS spectroscopies. 16th International Conference on X-ray absorption fine structure (XAFS16); Journal of physics conference series 2016; 712: 1-4
- 7 Jara GE, Solis CA, Gspooner NS. , et al. An experimental and TD-DFT theoretical study on the photophysical properties of methylene violet (Bernthsen). Dyes and Pigments 2015; 112: 341-351
- 8 Ronzani F, Trivella A, Bordat P. , et al. Revisiting the photophysics and photochemistry of methylene violet (MV). J Photochem Photobiol Chem 2014; 284: 8-17
- 9 Davis F, Higson S. Cyclodextrins. In Macrocycles Construction, Chemistry and Nanotechnology Applications. New York, NY: Wiley; 2011: 190-244
- 10 Holt JS, Campitella A, Rich A, Young JL. Spectroscopic characterization of the binding and isomerization cycle of merocyanine with α-, β-, and γ-cyclodextrins. J Incl Phenom Macrocycl Chem 2008; 61: 251-258
- 11 Steiner U, Abdel-Kader MH, Fischer P, Kramer HEA. Photochemical cis/trans isomerisation of a stilbazolium betaine. A protolytic/photochemical reaction cycle. J Am Chem Soc 1978; 10: 3190-3197
- 12 Gonzalez D, Neilands O, Rezende MC. The solvatochromic behaviour of 2- and 4-pyridiniophenoxides. J Chem Soc, Perkin Trans 2 1999; 4: 713-717
- 13 Albert A, Sergeant EP. Zwitterions (Dipolar Ions). In The Determination of Ionization Constants. London: Chapman and Hall; 1971: 76-81
- 14 Perrin DD, Dempsey B, Serjeant EP. pKa Prediction for Organic Acids and Bases. London: Chapman and Hall; 1981
- 15 Davis F, Higson S. Cucurbiturils. In Macrocycles: Construction, Chemistry and Nanotechnology Applications. New York, NY: Wiley; 2011: 325-368
- 16 Minkin VI, Osipov OA, Zhdanov YA. Dipole Moments in Organic Chemistry. New York, NY: Springer; 1970
- 17 Niewodniczański W, Bartkowiak W. Theoretical study of geometrical and nonlinear optical properties of pyridinium N-phenolate betaine dyes. J Mol Model 2007; 13: 793-800
- 18 Wyn-Jones J, Gormally J. Aggregation Processes in Solution (Studies in Physical and Theoretical Chemistry 26). Amsterdam: Elsevier; 1983
- 19 Hahnemann S. Organon of Medicine. 5th and 6th eds
- 20 Meichsner J, Schmidt M, Schneider R, Wagner HE. Nonthermal Plasma: Chemistry and Physics. Boca Raton: CRC Press; 2013
- 21 Nakayama K. Triboemission, triboplasma generation, and tribochemistry. In: Wang QJ, Chung YW. , eds. Encyclopedia of Tribology. New York, NY: Springer; 2013: 3750-3760
- 22 Nikitenk SI. Plasma formation during acoustic cavitation: toward a new paradigm for sonochemistry. Adv Phys Chem 2014; 2014: 173878 http://dx.doi.org/10.1155/2014/173878
- 23 Hibou F. Could the study of cavitation luminescence be useful in high dilution research?. Homeopathy 2017; 106: 181-190
Address for correspondence
Steven J. Cartwright, PhD
DiagnOx Laboratory, Cherwell Innovation Centre
Upper Heyford, Oxon, OX25 5HD
United Kingdom
Email: [email protected]



